BGEN.jl
Routines for reading compressed storage of genotyped or imputed markers
The BGEN Format
Genome-wide association studies (GWAS) data with imputed markers are often saved in the BGEN format or .bgen file.
Used in:
- Wellcome Trust Case-Control Consortium 2
- the MalariaGEN project
- the ALSPAC study
- UK Biobank: for genome-wide imputed genotypes and phased haplotypes
Features
- Can store both hard-calls and imputed data
- Can store both phased haplotypes and phased genotypes
- Efficient variable-precision bit reapresntations
- Per-variant compression $\rightarrow$ easy to index
Time to list variant identifying information (genomic location, ID and alleles): 18,496 samples, 121,668 SNPs (image source: https://www.well.ox.ac.uk/~gav/bgenformat/images/bgencomparison.png) 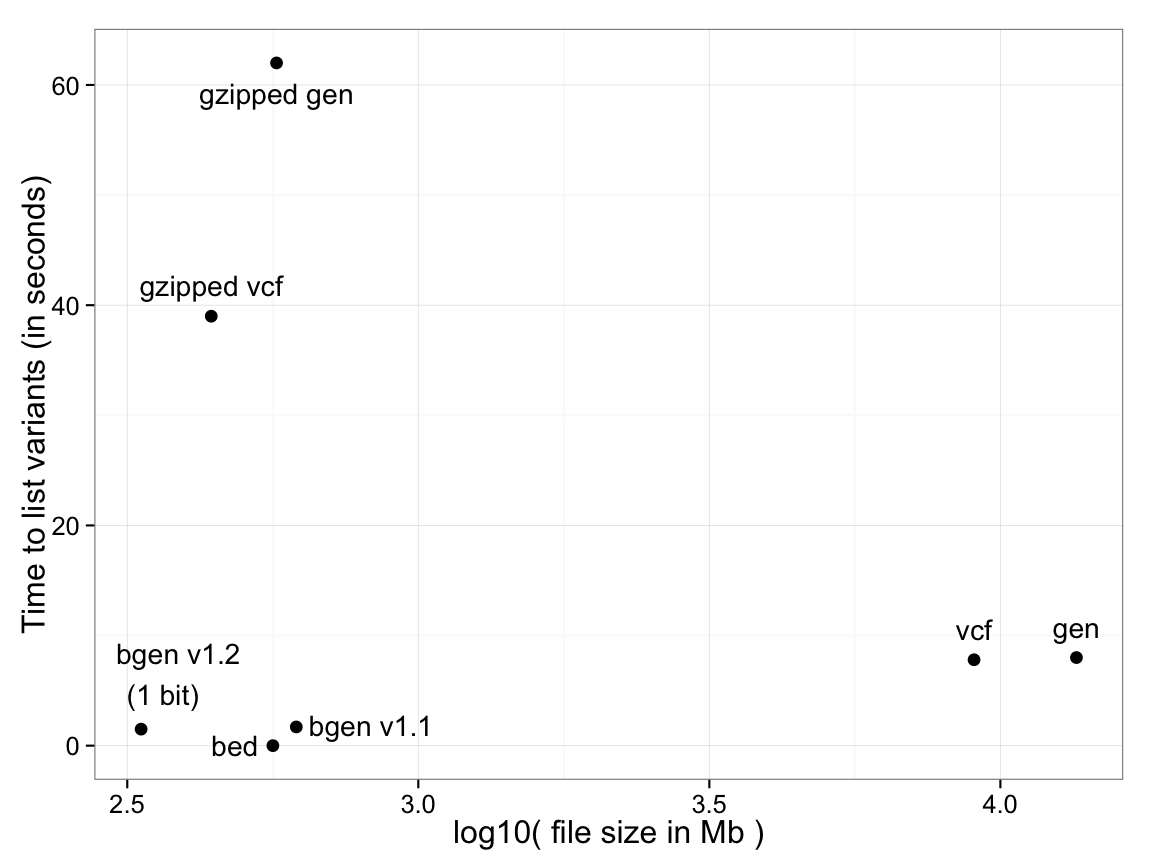
Plink 1 format (.bed/.bim/.fam) has the list of variants as a separate file (.bim), effectively zero time.
Structure
A header block followed by a series of [variant data block - (compressed) genotype data block] pairs.
- Header block
- number of variants and samples
- compression method (none, zlib or zstandard)
- version of layout
- Only "layout 2" is discussed below. "Layout 1" is also supported.
- sample identifiers (optional)
- Variant data block
- variant id
- genomic position (chromosome, bp coordinate)
- list of alleles
- Genotype data block (often compressed)
- ploidy of each sample (may vary sample-by-sample)
- if the genotype data are phased
- precision ($B$, number of bits to represent probabilities)
- probabilitiy data (e.g. an unsigned $B$-bit integer $x$ represents the probability of ($\frac{x}{2^{B}-1}$)
BGEN.jl provides tools for iterating over the variants and parsing genotype data efficiently. It has been optimized for UK Biobank's zlib-compressed, 8-bit byte-aligned, all-diploid, all-biallelic datafiles.
Installation
This package requires Julia v1.0 or later, which can be obtained from https://julialang.org/downloads/ or by building Julia from the sources in the https://github.com/JuliaLang/julia repository.
The package can be installed by running the following code:
using Pkg
pkg"add BGEN"In order to run the examples below, the Glob package is also needed.
pkg"add Glob"versioninfo()Julia Version 1.8.5
Commit 17cfb8e65ea (2023-01-08 06:45 UTC)
Platform Info:
OS: macOS (arm64-apple-darwin21.5.0)
CPU: 8 × Apple M2
WORD_SIZE: 64
LIBM: libopenlibm
LLVM: libLLVM-13.0.1 (ORCJIT, apple-m1)
Threads: 1 on 4 virtual coresusing BGEN, GlobExample Data
The example datafiles are stored in /data directory of this repository. It can be accessed through the function BGEN.datadir(). These files come from the reference implementation for the BGEN format.
Glob.glob("*", BGEN.datadir())79-element Vector{String}:
"/Users/kose/.julia/dev/BGEN/src/../data/LICENSE.md"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.10bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.11bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.12bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.13bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.14bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.15bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.16bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.17bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.18bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.19bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.1bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/complex.20bits.bgen"
⋮
"/Users/kose/.julia/dev/BGEN/src/../data/example.6bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/example.7bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen.bgi"
"/Users/kose/.julia/dev/BGEN/src/../data/example.9bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/example.gen"
"/Users/kose/.julia/dev/BGEN/src/../data/example.sample"
"/Users/kose/.julia/dev/BGEN/src/../data/example.v11.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/examples.16bits.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/haplotypes.bgen"
"/Users/kose/.julia/dev/BGEN/src/../data/haplotypes.bgen.bgi"
"/Users/kose/.julia/dev/BGEN/src/../data/haplotypes.haps"There are three different datasets with different format versions, compressions, or number of bits to represent probability values.
example.*.bgen: imputed genotypes.haplotypes.bgen: phased haplotypes.complex.*.bgen: includes imputed genotypes and phased haplotypes, and multiallelic genotypes.
Some of the .bgen files are indexed with .bgen.bgi files:
Glob.glob("*.bgen.bgi", BGEN.datadir())4-element Vector{String}:
"/Users/kose/.julia/dev/BGEN/src/../data/complex.bgen.bgi"
"/Users/kose/.julia/dev/BGEN/src/../data/example.16bits.bgen.bgi"
"/Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen.bgi"
"/Users/kose/.julia/dev/BGEN/src/../data/haplotypes.bgen.bgi"Sample identifiers may be either contained in the .bgen file, or is listed in an external .sample file.
Glob.glob("*.sample", BGEN.datadir())2-element Vector{String}:
"/Users/kose/.julia/dev/BGEN/src/../data/complex.sample"
"/Users/kose/.julia/dev/BGEN/src/../data/example.sample"Type Bgen
The type Bgen is the fundamental type for .bgen-formatted files. It can be created using the following line.
b = Bgen(BGEN.datadir("example.8bits.bgen");
sample_path=BGEN.datadir("example.sample"),
idx_path=BGEN.datadir("example.8bits.bgen.bgi"))Bgen(IOStream(<file /Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen>), 0x000000000001f6ea, BGEN.Header(0x0000178c, 0x00000014, 0x000000c7, 0x000001f4, 0x01, 0x02, true), ["sample_001", "sample_002", "sample_003", "sample_004", "sample_005", "sample_006", "sample_007", "sample_008", "sample_009", "sample_010" … "sample_491", "sample_492", "sample_493", "sample_494", "sample_495", "sample_496", "sample_497", "sample_498", "sample_499", "sample_500"], Index("/Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen.bgi", SQLite.DB("/Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen.bgi"), UInt64[], String[], String[], UInt32[]))The first argument is the path to the .bgen file. The optional keyword argument sample_path defines the location of the .sample file. The second optional keyword argument idx_path determines the location of .bgen.bgi file.
When a Bgen object is created, information in the header is parsed, and the index files are loaded if provided. You may retrieve basic information as follows. Variants are not yet parsed, and will be discussed later.
io(b::Bgen): IOStream for the bgen file. You may also close this stream usingclose(b::Bgen).fsize(b::Bgen): the size of the bgen file.samples(b::Bgen): the list of sample names.n_samples(b::Bgen): number of samples in the file.n_variants(b::Bgen): number of variantscompression(b::Bgen): the method each genotype block is compressed. It is either "None", "Zlib", or "Zstd".
io(b)IOStream(<file /Users/kose/.julia/dev/BGEN/src/../data/example.8bits.bgen>)fsize(b)128746samples(b)500-element Vector{String}:
"sample_001"
"sample_002"
"sample_003"
"sample_004"
"sample_005"
"sample_006"
"sample_007"
"sample_008"
"sample_009"
"sample_010"
"sample_011"
"sample_012"
"sample_013"
⋮
"sample_489"
"sample_490"
"sample_491"
"sample_492"
"sample_493"
"sample_494"
"sample_495"
"sample_496"
"sample_497"
"sample_498"
"sample_499"
"sample_500"n_samples(b)500n_variants(b)199compression(b)"Zlib"One may also access the list of RSIDs, chromosomes, and positions in chromosome of each variant stored using functions rsids(), chroms(), and positions(), respectively.
rsids(b)199-element Vector{String}:
"RSID_101"
"RSID_2"
"RSID_102"
"RSID_3"
"RSID_103"
"RSID_4"
"RSID_104"
"RSID_5"
"RSID_105"
"RSID_6"
"RSID_106"
"RSID_7"
"RSID_107"
⋮
"RSID_194"
"RSID_95"
"RSID_195"
"RSID_96"
"RSID_196"
"RSID_97"
"RSID_197"
"RSID_98"
"RSID_198"
"RSID_99"
"RSID_199"
"RSID_200"chroms(b)199-element Vector{String}:
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
⋮
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"
"01"positions(b)199-element Vector{Int64}:
1001
2000
2001
3000
3001
4000
4001
5000
5001
6000
6001
7000
7001
⋮
94001
95000
95001
96000
96001
97000
97001
98000
98001
99000
99001
100001Variant and VariantIterator
As noted earlier, genotype information of each variant is compressed separately in .bgen files. The offsets (starting points in bgen file) of the genotypes may or may not be indexed by an external .bgen.bgi file. Thus, two ways to iterate over variants is provided through the function iterator(b; offsets=nothing, from_bgen_start=false).
- If
offsetsis provided, or.bgen.bgiis provided and
from_bgen_start is false, it returns a VariantIteratorFromOffsets, iterating over the list of offsets.
- Otherwise, it returns a
VariantIteratorFromStart, iterating from the start of bgen file to the end of it sequentially.
VariantIteratorFromOffsets and VariantIteratorFromStart are the subtypes of VariantIterator.
Each element of VariantIterator is a Variant, containing the information of variants. We have following utility functions to access its information.
n_samples(v::Variant)varid(v::Variant)rsid(v::Variant)chrom(v::Variant)pos(v::Variant)n_alleles(v::Variant): number of alleles.alleles(v::Variant): list of alleles.
Merely the basic information of a variant is parsed for creating a Variant object. Nothing is decompressed, and genotype probabilities are not yet parsed yet. Decompression happens lazily, and is delayed until when we try to compute genotype probabilites or minor allele dosages (to be discussed later).
Since .bgen.bgi file is provided, the following order is based on the index file, sorted by genomic location.
for v in iterator(b) #
println(rsid(v))
endRSID_101
RSID_2
RSID_102
RSID_3
RSID_103
RSID_4
RSID_104
RSID_5
RSID_105
RSID_6
RSID_106
RSID_7
RSID_107
RSID_8
RSID_108
RSID_9
RSID_109
RSID_10
RSID_100
RSID_110
RSID_11
RSID_111
RSID_12
RSID_112
RSID_13
RSID_113
RSID_14
RSID_114
RSID_15
RSID_115
RSID_16
RSID_116
RSID_17
RSID_117
RSID_18
RSID_118
RSID_19
RSID_119
RSID_20
RSID_120
RSID_21
RSID_121
RSID_22
RSID_122
RSID_23
RSID_123
RSID_24
RSID_124
RSID_25
RSID_125
RSID_26
RSID_126
RSID_27
RSID_127
RSID_28
RSID_128
RSID_29
RSID_129
RSID_30
RSID_130
RSID_31
RSID_131
RSID_32
RSID_132
RSID_33
RSID_133
RSID_34
RSID_134
RSID_35
RSID_135
RSID_36
RSID_136
RSID_37
RSID_137
RSID_38
RSID_138
RSID_39
RSID_139
RSID_40
RSID_140
RSID_41
RSID_141
RSID_42
RSID_142
RSID_43
RSID_143
RSID_44
RSID_144
RSID_45
RSID_145
RSID_46
RSID_146
RSID_47
RSID_147
RSID_48
RSID_148
RSID_49
RSID_149
RSID_50
RSID_150
RSID_51
RSID_151
RSID_52
RSID_152
RSID_53
RSID_153
RSID_54
RSID_154
RSID_55
RSID_155
RSID_56
RSID_156
RSID_57
RSID_157
RSID_58
RSID_158
RSID_59
RSID_159
RSID_60
RSID_160
RSID_61
RSID_161
RSID_62
RSID_162
RSID_63
RSID_163
RSID_64
RSID_164
RSID_65
RSID_165
RSID_66
RSID_166
RSID_67
RSID_167
RSID_68
RSID_168
RSID_69
RSID_169
RSID_70
RSID_170
RSID_71
RSID_171
RSID_72
RSID_172
RSID_73
RSID_173
RSID_74
RSID_174
RSID_75
RSID_175
RSID_76
RSID_176
RSID_77
RSID_177
RSID_78
RSID_178
RSID_79
RSID_179
RSID_80
RSID_180
RSID_81
RSID_181
RSID_82
RSID_182
RSID_83
RSID_183
RSID_84
RSID_184
RSID_85
RSID_185
RSID_86
RSID_186
RSID_87
RSID_187
RSID_88
RSID_188
RSID_89
RSID_189
RSID_90
RSID_190
RSID_91
RSID_191
RSID_92
RSID_192
RSID_93
RSID_193
RSID_94
RSID_194
RSID_95
RSID_195
RSID_96
RSID_196
RSID_97
RSID_197
RSID_98
RSID_198
RSID_99
RSID_199
RSID_200Setting from_bgen_start=true forces the iterator to iterate in the order of appearence in the bgen file. This may be different from the order in the index file.
for v in iterator(b; from_bgen_start=true)
println(rsid(v))
endRSID_2
RSID_3
RSID_4
RSID_5
RSID_6
RSID_7
RSID_8
RSID_9
RSID_10
RSID_11
RSID_12
RSID_13
RSID_14
RSID_15
RSID_16
RSID_17
RSID_18
RSID_19
RSID_20
RSID_21
RSID_22
RSID_23
RSID_24
RSID_25
RSID_26
RSID_27
RSID_28
RSID_29
RSID_30
RSID_31
RSID_32
RSID_33
RSID_34
RSID_35
RSID_36
RSID_37
RSID_38
RSID_39
RSID_40
RSID_41
RSID_42
RSID_43
RSID_44
RSID_45
RSID_46
RSID_47
RSID_48
RSID_49
RSID_50
RSID_51
RSID_52
RSID_53
RSID_54
RSID_55
RSID_56
RSID_57
RSID_58
RSID_59
RSID_60
RSID_61
RSID_62
RSID_63
RSID_64
RSID_65
RSID_66
RSID_67
RSID_68
RSID_69
RSID_70
RSID_71
RSID_72
RSID_73
RSID_74
RSID_75
RSID_76
RSID_77
RSID_78
RSID_79
RSID_80
RSID_81
RSID_82
RSID_83
RSID_84
RSID_85
RSID_86
RSID_87
RSID_88
RSID_89
RSID_90
RSID_91
RSID_92
RSID_93
RSID_94
RSID_95
RSID_96
RSID_97
RSID_98
RSID_99
RSID_100
RSID_101
RSID_102
RSID_103
RSID_104
RSID_105
RSID_106
RSID_107
RSID_108
RSID_109
RSID_110
RSID_111
RSID_112
RSID_113
RSID_114
RSID_115
RSID_116
RSID_117
RSID_118
RSID_119
RSID_120
RSID_121
RSID_122
RSID_123
RSID_124
RSID_125
RSID_126
RSID_127
RSID_128
RSID_129
RSID_130
RSID_131
RSID_132
RSID_133
RSID_134
RSID_135
RSID_136
RSID_137
RSID_138
RSID_139
RSID_140
RSID_141
RSID_142
RSID_143
RSID_144
RSID_145
RSID_146
RSID_147
RSID_148
RSID_149
RSID_150
RSID_151
RSID_152
RSID_153
RSID_154
RSID_155
RSID_156
RSID_157
RSID_158
RSID_159
RSID_160
RSID_161
RSID_162
RSID_163
RSID_164
RSID_165
RSID_166
RSID_167
RSID_168
RSID_169
RSID_170
RSID_171
RSID_172
RSID_173
RSID_174
RSID_175
RSID_176
RSID_177
RSID_178
RSID_179
RSID_180
RSID_181
RSID_182
RSID_183
RSID_184
RSID_185
RSID_186
RSID_187
RSID_188
RSID_189
RSID_190
RSID_191
RSID_192
RSID_193
RSID_194
RSID_195
RSID_196
RSID_197
RSID_198
RSID_199
RSID_200With the presence of .bgen.bgi index file, one may select variants on a certain region using the function select_region(b, chrom; start=nothing, stop=nothing).
The following shows that all 199 variants in the bgen file are located on chromosome 01.
length(select_region(b, "01"))199We can see that the first variant since position 5000 at chromosome 01 is "RSID_5":
first(select_region(b, "01"; start=5000))Variant(0x0000000000001ef8, 0x0000000000001f21, 0x0000000000002169, 0x00000248, 0x000001f4, "SNPID_5", "RSID_5", "01", 0x00001388, 0x0002, ["A", "G"], nothing)And that the number of variants in chr01:5000-50000 is 92.
length(select_region(b, "01"; start=5000, stop=50000))92Finally, one may use the parse_variants() function to retrieve the variant information as a Vector{Variant}. This is equivalent to calling collect() on the corresponding VariantIterator. It takes the same arguments as iterator(). This keeps all the information of variants in-memory. If the size of bgen file is too large, you might want to avoid this.
variants = parse_variants(b; from_bgen_start=true)199-element Vector{Variant}:
Variant(0x0000000000001790, 0x00000000000017b9, 0x0000000000001a82, 0x000002c9, 0x000001f4, "SNPID_2", "RSID_2", "01", 0x000007d0, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000001a82, 0x0000000000001aab, 0x0000000000001ced, 0x00000242, 0x000001f4, "SNPID_3", "RSID_3", "01", 0x00000bb8, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000001ced, 0x0000000000001d16, 0x0000000000001ef8, 0x000001e2, 0x000001f4, "SNPID_4", "RSID_4", "01", 0x00000fa0, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000001ef8, 0x0000000000001f21, 0x0000000000002169, 0x00000248, 0x000001f4, "SNPID_5", "RSID_5", "01", 0x00001388, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000002169, 0x0000000000002192, 0x0000000000002389, 0x000001f7, 0x000001f4, "SNPID_6", "RSID_6", "01", 0x00001770, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000002389, 0x00000000000023b2, 0x00000000000025df, 0x0000022d, 0x000001f4, "SNPID_7", "RSID_7", "01", 0x00001b58, 0x0002, ["A", "G"], nothing)
Variant(0x00000000000025df, 0x0000000000002608, 0x00000000000027a4, 0x0000019c, 0x000001f4, "SNPID_8", "RSID_8", "01", 0x00001f40, 0x0002, ["A", "G"], nothing)
Variant(0x00000000000027a4, 0x00000000000027cd, 0x00000000000029de, 0x00000211, 0x000001f4, "SNPID_9", "RSID_9", "01", 0x00002328, 0x0002, ["A", "G"], nothing)
Variant(0x00000000000029de, 0x0000000000002a09, 0x0000000000002c43, 0x0000023a, 0x000001f4, "SNPID_10", "RSID_10", "01", 0x00002710, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000002c43, 0x0000000000002c6e, 0x0000000000002e8a, 0x0000021c, 0x000001f4, "SNPID_11", "RSID_11", "01", 0x00002af8, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000002e8a, 0x0000000000002eb5, 0x00000000000030e0, 0x0000022b, 0x000001f4, "SNPID_12", "RSID_12", "01", 0x00002ee0, 0x0002, ["A", "G"], nothing)
Variant(0x00000000000030e0, 0x000000000000310b, 0x0000000000003375, 0x0000026a, 0x000001f4, "SNPID_13", "RSID_13", "01", 0x000032c8, 0x0002, ["A", "G"], nothing)
Variant(0x0000000000003375, 0x00000000000033a0, 0x00000000000035dd, 0x0000023d, 0x000001f4, "SNPID_14", "RSID_14", "01", 0x000036b0, 0x0002, ["A", "G"], nothing)
⋮
Variant(0x000000000001d991, 0x000000000001d9be, 0x000000000001dc12, 0x00000254, 0x000001f4, "SNPID_189", "RSID_189", "01", 0x00015ba9, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001dc12, 0x000000000001dc3f, 0x000000000001ddf2, 0x000001b3, 0x000001f4, "SNPID_190", "RSID_190", "01", 0x00015f91, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001ddf2, 0x000000000001de1f, 0x000000000001e011, 0x000001f2, 0x000001f4, "SNPID_191", "RSID_191", "01", 0x00016379, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001e011, 0x000000000001e03e, 0x000000000001e214, 0x000001d6, 0x000001f4, "SNPID_192", "RSID_192", "01", 0x00016761, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001e214, 0x000000000001e241, 0x000000000001e407, 0x000001c6, 0x000001f4, "SNPID_193", "RSID_193", "01", 0x00016b49, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001e407, 0x000000000001e434, 0x000000000001e6c9, 0x00000295, 0x000001f4, "SNPID_194", "RSID_194", "01", 0x00016f31, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001e6c9, 0x000000000001e6f6, 0x000000000001e8e1, 0x000001eb, 0x000001f4, "SNPID_195", "RSID_195", "01", 0x00017319, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001e8e1, 0x000000000001e90e, 0x000000000001ec86, 0x00000378, 0x000001f4, "SNPID_196", "RSID_196", "01", 0x00017701, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001ec86, 0x000000000001ecb3, 0x000000000001ef8b, 0x000002d8, 0x000001f4, "SNPID_197", "RSID_197", "01", 0x00017ae9, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001ef8b, 0x000000000001efb8, 0x000000000001f183, 0x000001cb, 0x000001f4, "SNPID_198", "RSID_198", "01", 0x00017ed1, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001f183, 0x000000000001f1b0, 0x000000000001f3d4, 0x00000224, 0x000001f4, "SNPID_199", "RSID_199", "01", 0x000182b9, 0x0002, ["A", "G"], nothing)
Variant(0x000000000001f3d4, 0x000000000001f401, 0x000000000001f6ea, 0x000002e9, 0x000001f4, "SNPID_200", "RSID_200", "01", 0x000186a1, 0x0002, ["A", "G"], nothing)If the index file (.bgi) is provided, the users may search for certain RSID in a BGEN file.
v = variant_by_rsid(b, "RSID_10")Variant(0x00000000000029de, 0x0000000000002a09, 0x0000000000002c43, 0x0000023a, 0x000001f4, "SNPID_10", "RSID_10", "01", 0x00002710, 0x0002, ["A", "G"], nothing)Also, the users may look for the n-th (1-based) variant with respect to genomic location.
v = variant_by_index(b, 4)Variant(0x0000000000001a82, 0x0000000000001aab, 0x0000000000001ced, 0x00000242, 0x000001f4, "SNPID_3", "RSID_3", "01", 0x00000bb8, 0x0002, ["A", "G"], nothing)Genotype/haplotype probabilities and minor allele dosage
The genotype information is decompressed and parsed when probability data is needed. The parsing is triggered by a call to one of:
probabilities!(b::Bgen, v::Variant; T=Float64): probability of each genotype/haplotype.first_allele_dosage!(b::Bgen, v::Variant; T=Float64) : dosage of the first allele for a biallelic variant. The first allele listed is often the alternative allele, but it depends on project-wise convention. For example, the first allele is the reference allele for the UK Biobank project.minor_allele_dosage!(b::Bgen, v::Variant; T=Float64): minor allele dosage for a biallelic variant.
Once parsed, the results are cached and loaded on any subsequent calls. After that, one may access genotype information using the following functions, as well as probabilities!() and minor_allele_dosage!():
phased(v::Variant): if the stored data is phasedmin_ploidy(v::Variant): minimum ploidy across the samplesmax_ploidy(v::Variant): maximum ploidy across the samplesploidy(v::Variant): Vector of ploidy for each samplebit_depth(v::Variant): number of bits used to represent a probability valuemissings(v::Variant): list of samples data is missing
These functions are allowed after calling minor_allele_dosage!():
minor_allele(v::Variant)major_allele(v::Variant)
If the data are not phased, probabilities!(b, v)[i, j] represents the probability of genotype i for sample j. Each column sums up to one. The genotypes are in colex-order of allele counts. For example, for three alleles with ploidy 3:
| row index | allele counts | genotype |
|---|---|---|
| 1 | (3, 0, 0) | 111 |
| 2 | (2, 1, 0) | 112 |
| 3 | (1, 2, 0) | 122 |
| 4 | (0, 3, 0) | 222 |
| 5 | (2, 0, 1) | 113 |
| 6 | (1, 1, 1) | 123 |
| 7 | (0, 2, 1) | 223 |
| 8 | (1, 0, 2) | 133 |
| 9 | (0, 1, 2) | 233 |
| 10 | (0, 0, 3) | 333 |
probabilities!(b, variants[1])3×500 Matrix{Float32}:
NaN 0.027451 0.0156863 0.0235294 … 0.0156863 0.921569 0.00392157
NaN 0.00784314 0.0509804 0.933333 0.027451 0.0509804 0.984314
NaN 0.964706 0.933333 0.0431373 0.956863 0.027451 0.0117647Genotype data for sample 1 is missing in this case.
missings(variants[1])1-element Vector{Int64}:
1On the other hand, if the data are phased, probabilities!(b, v)[i, j] represents the probability that haplotype (i - 1) ÷ n_alleles + 1 has allele (i - 1) % n_alleles + 1 for sample j, where n_alleles is the number of alleles. The below is an example of phased probabilities. i.e., each column represents each sample, and each group of n_alleles rows represent the allele probabilities for each haplotype. In this case, ploidy is [1, 2, 2, 2], thus indexes [3:4, 1] are invalid, and is filled with NaN.
b2 = Bgen(BGEN.datadir("complex.bgen"))
vs = parse_variants(b2)
p = probabilities!(b2, vs[3])4×4 Matrix{Float32}:
1.0 0.0 1.0 1.0
0.0 1.0 0.0 0.0
NaN 0.0 1.0 0.0
NaN 1.0 0.0 1.0This variant has two possible alleles (allele 1: "A" and allele 2: "G"), and all the samples are diploids except for the first one, which is monoploid.
It corresponds to a line of VCF file:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample_0 sample_1 sample_2 sample_3
01 3 V3 A G . . . GT:HP 0:1,0 1|1:0,1,0,1 0|0:1,0,1,0 0|1:1,0,0,1So the first sample is monoploid of A, the second sample is homozygote A|A, the third sample is homozygote G|G, and the last sample is heterozygote A|G (phased).
We can confirm the phasedness and ploidy of each sample as follows.
phased(vs[3])0x01ploidy(vs[3])4-element Vector{UInt8}:
0x01
0x02
0x02
0x02alleles(vs[3])2-element Vector{String}:
"A"
"G"For biallelic genotype data, first_allele_dosage!(b, v) and minor_allele_dosage!(b, v) can be computed. It also supports phased data.
first_allele_dosage!(b, variants[1])500-element Vector{Float32}:
NaN
0.0627451
0.08235294
0.9803922
0.09019608
0.14117648
1.0745099
0.054901965
0.10980393
0.121568635
0.14117648
0.21568629
0.08235294
⋮
0.09411766
0.10196079
0.027450982
0.96470594
0.0
1.0117648
0.043137256
0.0627451
1.0431373
0.05882353
1.8941176
0.99215686minor_allele_dosage!(b, variants[1])500-element Vector{Float32}:
NaN
0.0627451
0.08235294
0.9803922
0.09019608
0.14117648
1.0745099
0.054901965
0.10980393
0.121568635
0.14117648
0.21568629
0.08235294
⋮
0.09411766
0.10196079
0.027450982
0.96470594
0.0
1.0117648
0.043137256
0.0627451
1.0431373
0.05882353
1.8941176
0.99215686phased(variants[1])0x00n_alleles(variants[1])2minor_allele(variants[1])"A"major_allele(variants[1])"G"first_allele_dosage!() and minor_allele_dosage!() support a keyword argument mean_impute, which imputes missing value with the mean of the non-missing values.
dose = first_allele_dosage!(b, variants[1]; T=Float64, mean_impute=true)500-element Vector{Float32}:
0.39581063
0.0627451
0.08235294
0.9803922
0.09019608
0.14117648
1.0745099
0.054901965
0.10980393
0.121568635
0.14117648
0.21568629
0.08235294
⋮
0.09411766
0.10196079
0.027450982
0.96470594
0.0
1.0117648
0.043137256
0.0627451
1.0431373
0.05882353
1.8941176
0.99215686The function hardcall(d; threshold=0.1) can be used to convert the dosage vectors to hard-called genotypes, returning a Vector{UInt8} array of values 0x00, 0x01, 0x02, or 0x09 (for missing). The function hardcall!(c, d; threshold=0.1) can be used to fill in a preallocated integer array c. threshold determines the maximum distance between the hard genotypes and the dosage values. For example, if threshold = 0.1, dosage value in [0, 0.1) gives the hard call 0x00, a value in (0.9, 1,1) gives 0x01, and a value in (1.9, 2.0] gives 0x02. Any other values give 0x09.
c = hardcall(dose; threshold=0.1)500-element Vector{UInt8}:
0x09
0x00
0x00
0x01
0x00
0x09
0x01
0x00
0x09
0x09
0x09
0x09
0x00
⋮
0x00
0x09
0x00
0x01
0x00
0x01
0x00
0x00
0x01
0x00
0x09
0x01calls = Vector{UInt8}(undef, length(dose))
hardcall!(calls, dose; threshold = 0.1)500-element Vector{UInt8}:
0x09
0x00
0x00
0x01
0x00
0x09
0x01
0x00
0x09
0x09
0x09
0x09
0x00
⋮
0x00
0x09
0x00
0x01
0x00
0x01
0x00
0x00
0x01
0x00
0x09
0x01Filtering
Filtering based on BitVector for samples and variants is supported through BGEN.filter function if bit depth of each variant is a multiple of 8. The syntax is:
BGEN.filter(dest::AbstractString, b::Bgen, variant_mask::BitVector,
sample_mask::BitVector=trues(length(b.samples));
dest_sample = dest[1:end-5] * ".sample",
sample_path=nothing, sample_names=b.samples,
offsets=nothing, from_bgen_start=false)destis the output path of the resulting.bgenfile.bis aBgeninstance.variant_maskis aBitVectorfor determining whether to include each variant in the output file.sample_maskis aBitVectorfor determining whether to include each sample in the output file.dest_sampleis the location of the output.samplefile.sample_pathis the location of the.samplefile of the input BGEN file.sample_namesis the names of the sample in the input BGEN file.offsetsandfrom_bgen_startare the arguments for theiteratormethod.
It only supports layout 2, and the output is always compressed in ZSTD. The sample names are stored in a separate .sample file, but not in the output .bgen file.
An example of choosing first 10 variants:
b = Bgen(BGEN.datadir("example.8bits.bgen"))
vidx = falses(b.header.n_variants)
vidx[1:10] .= true
BGEN.filter("test.bgen", b, vidx)
b2 = Bgen("test.bgen"; sample_path="test.sample")
@assert all(b.samples .== b2.samples)
for (v1, v2) in zip(iterator(b), iterator(b2)) # length of two iterators are different.
# it stops when the shorter one (b2) ends.
@assert v1.varid == v2.varid
@assert v1.rsid == v2.rsid
@assert v1.chrom == v2.chrom
@assert v1.pos == v2.pos
@assert v1.n_alleles == v2.n_alleles
@assert all(v1.alleles .== v2.alleles)
decompressed1 = BGEN.decompress(b.io, v1, b.header)
decompressed2 = BGEN.decompress(b2.io, v2, b2.header)
@assert all(decompressed1 .== decompressed2)
endAn example of choosing last two samples out of four samples:
b3 = Bgen(BGEN.datadir("complex.24bits.bgen"))
BGEN.filter("test2.bgen", b3, trues(b3.header.n_variants), BitVector([false, false, true, true]))
b4 = Bgen("test2.bgen"; sample_path = "test2.sample")
for (v3, v4) in zip(iterator(b3), iterator(b4))
@assert v3.varid == v4.varid
@assert v3.rsid == v4.rsid
@assert v3.chrom == v4.chrom
@assert v3.pos == v4.pos
@assert v3.n_alleles == v4.n_alleles
@assert all(v3.alleles .== v4.alleles)
@assert isapprox(probabilities!(b3, v3)[:, 3:4], probabilities!(b4, v4); nans=true)
endrm("test.bgen", force=true)
rm("test.sample", force=true)
rm("test2.bgen", force=true)
rm("test2.sample", force=true)